Usage¶
Alvis can be run from the command line using the .jar file. In the terminal, navigate to the dist directory in the Alvis project, and type e.g:
Java -jar Alvis.jar -type alignment -inputfmt paf -outputfmt tex \
-in /path/to/alignments/alignments.paf -outdir /path/to/out/ -out prefix
This will create the file prefix_alignment.tex in the directory /path/to/out. The file prefix_alignment.tex can then be compiled by your favourite tex compiler and viewed as a pdf. The options used in this command form the minimum set required for Alvis to work, and are described in the following table.
| Option <argument> | Description |
|---|---|
-type <diagram> |
Type of diagram to be produced. |
-inputfmt <format> |
Format of the alignment file to use. |
-outputfmt <format> |
Format of the output diagram (default: tex). |
-in <path> |
Full file path to alignment file. |
-outdir <dir> |
Directory to write output to. Alvis will create this directory if it does not exist (default: ./alvis_output). |
-out <prefix> |
Prefix to use for output file names (default: diagram). |
The following are optional.
| Option <argument> | Description |
|---|---|
-filter |
Filter small alignments to remove noise. |
-minAlignmentPC <int> |
Minimum size of alignments (as % of the query length) to keep when filtering. |
-chimeras |
Only display alignments from chimeras. |
-printChimeras |
Output a text file containing alignments belonging to chimeras. |
-chimeraPositions |
Include alignment start and end positions in
the file created by -printChimeras. |
-minChimeraCoveragePC <int> |
Minimum coverage of query (as % of the query length) for identifying chimeras. |
-minChimeraAlignmentPC <int> |
Minimum size of alignments (as % of the query length) for identifying chimeras. |
-alignmentQueryName <query sequence id> |
ID of query sequence for exapnded contig alignment diagram. |
-alignmentTargetName <target sequence id> |
ID of target sequence for expanded contig alignment diagram. |
-blastfmt <format string> |
Format string as given to blast. Must be
used if -inputfmt is blast. |
-tsizes <targets string> |
List of target names and sizes, separated by
whitespace. Required if inputfmt is
sam. |
-binsize <int> |
Size (in bp) of bins for genome coverage diagrams. |
-coverageType <type> |
The type of coverage heatmap to produce. Must be either “square” or “long”. |
-randomcolours <seed> |
Randomly generate reference sequence colours for contig alignment diagram, with seed. |
-colourfile <filename> |
Name of file specifying hex colour for each reference sequence. Each line must be in the format <ref sequence> <hex colour> |
Diagrams¶
The type of diagram is specified with the -type <diagram> option etc. The currently available diagrams are:
alignmentcontigAlignmentcoveragemapgenomecoverage
Detailed information for each diagram can be found in the Diagrams section.
Input Formats¶
Alvis accepts a variety of alignment formats. These are specified by the option -inputfmt <format>, where <format> is one of the following:
=
- paf
- psl
- blast
- coords
- tiling
- sam
Coords¶
A coords file can be created from a mummer .delta file using the show-coords command in the MUMer package. For this file to work with alvis, the -B option must be specified (see here for more details).
Tiling¶
Similarly, a tiling file can be created from a mummer .delta file using the show-tiling command. In this case, the -a option must be specified (see here for more details).
BLAST¶
If BLAST is given as the format, the input file must have been created by BLAST using the tabular option, and the following fields must be present in some order:
qseqidsseqidqstartqendqlensstartsendslen
The parameter passed to blast after the -outfmt option must also be given to Alvis after the -blastfmt option. For example, the following command could be used:
Java -jar Alvis.jar -type alignment -inputfmt blast -outputfmt tex \
-in /path/to/alignments/alignments.blast -outdir /path/to/out/ -out prefix \
-blastfmt '6 qseqid sseqid qstart qend qlen sstart ssend slen'
if the file alignment.blast was created with:
blastn -db nt -query query.fa -out alignments.blast -outfmt '6 qseqid sseqid qstart qend qlen sstart ssend slen'
SAM¶
When using a SAM file, Alvis will attempt to find the target contig sizes from the header section. If this unavailable, the user can supply these values through the -tsizes option, by typing a space-separaed list of target names and their sizes. E.g. -tsizes 'Chr1 34964571 Chr2 22037565 Chr3 25499034 Chr4 20862711 Chr5 31270811'.
Output Formats¶
Alvis can currently output most diagrams in two formats: SVG and laTeX. These are specified by the -outputfmt <format> option, where <format> is one of tex and svg. Note that if tex is specified, the user must compile the .tex file that is created to obtain a PDF. Currently the diagrams are drawn with the tikz library, so the user must have this installed.
The following table shows the accepted input and output formats for each diagram.
| Input Formats | Output Formats | |||||||
|---|---|---|---|---|---|---|---|---|
| blast | coords | tiling | paf | psl | sam | svg | tex | |
| Alignment Diagram | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Contig Alignment Diagram | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Coverage Map Diagram | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Genome Coverage Diagram | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
Filtering¶
The user can filter alignments using the -filter option. This will cause alvis to ignore all alignments with length less than -minAlignmentPC % of the reference contig size (set to 0.5% by default). Note that this option is currently only used by the alignment diagram and the contig alignment diagram.
When using the -chimera option in conjunction with the contig alignment diagram, alvis will display only those alignments that it thinks could be a chimera. These are chosen when a query sequence is at least 90% covered by exactly two non-overlapping alignments, either from different reference sequences, or different loci of the same reference sequence. Each of these alignments must have a length of at least 10% of the query sequence. These values may be adjusted by the user with the -minChimeraCoveragePC and -minChimeraAlignmentPC options. The user should be aware that sequences are assumed to be non-circular; chimeras may be found when a read covers the join of a circular sequence.
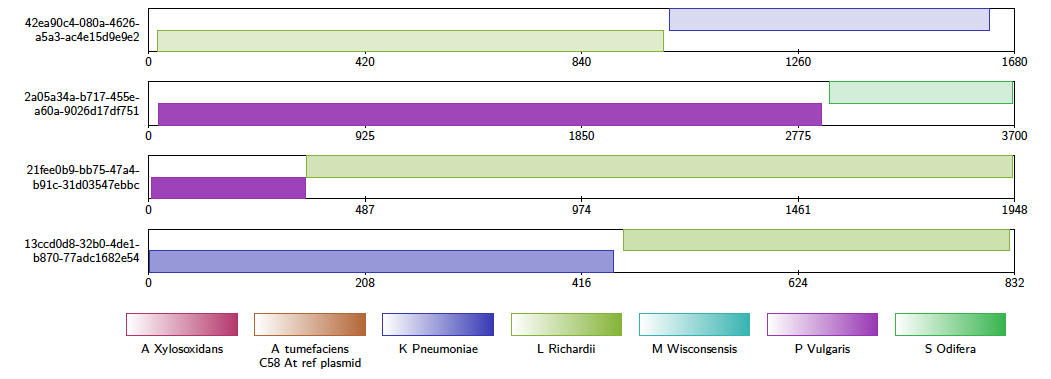
Additionally, when the -printChimeras option is specified, a text file named chimeras.txt is written to the output directory. This is a tab-seperated values file, where each line describes a potential chimera. Each line has the following fields.
| Column | Type | Description |
|---|---|---|
| 1 | String | Query sequence name. |
| 2 | int | Approximate position of chimera join on query sequence. |
| 3 | String | Target sequence name for first alignment. |
| 4 | String | Target sequence name for second alignment. |
If the option -chimeraPositions is also specified, then for each chimera, the start and end positions of the two alignments forming the chimera are also written to the file chimeras.txt, relative to both the query sequence (i.e. the read) and the target sequence (i.e. the reference). In this case, each line has the following fields.
| Column | Type | Description |
|---|---|---|
| 1 | String | Query sequence name. |
| 2 | int | Approximate position of chimera join on query sequence. |
| 3 | String | Target sequence name for first alignment. |
| 4 | String | Target sequence name for second alignment. |
| 5 | int | Start position on query of the first alignment. |
| 6 | int | End position on query of the first alignment. |
| 7 | int | Start position on query of the second alignment. |
| 8 | int | End position on query of the second alignment. |
| 9 | int | Start position on target of the first alignment. |
| 10 | int | End position on target of the first alignment. |
| 11 | int | Start position on target of the second alignment. |
| 12 | int | End position on target of the second alignment. |